
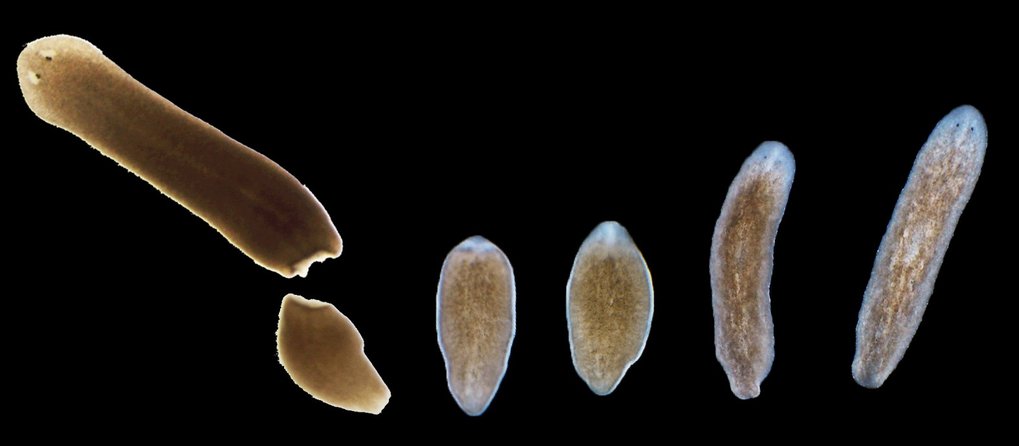
The planarian flatworm Schmidtea mediterranea is an extraordinary animal. Even when cut into tiny pieces, each piece can regenerate back into a complete and perfectly proportioned miniature planarian. Key to this ability are fascinating adult stem cells, a single one of which can restore a complete worm. But how Schmidtea mediterranea achieves these feats is so far poorly understood. An important step towards this goal is the first highly contiguous genome assembly ofSchmidtea mediterranea that researchers at the Max Planck Institute of Molecular Cell Biology and Genetics (MPI-CBG) in Dresden in cooperation with the Heidelberg Institute for Theoretical Studies (HITS) report in the current issue of Nature. The assembly reveals a genome that contains novel giant repeat elements, new flatworm-specific genes, but also the absence of other genes that were so far thought to be absolutely essential for keeping an animal alive. The discovery has potential implications in the fields of regeneration research, stem cell biology and bioinformatics.

The flatworm Schmidtea mediterranea can regenerate back into a complete organism from individual body parts. Researchers have now fully decoded this exceptionally repetitive genome, Credit: MPI for Molecular Cell Biology and Genetics / J. Rink
{kind=link}
A complete and fully assembled genome is critical for understanding the biological characteristics of an organism. Scientists have previously attempted to sequence the genome of Schmidtea mediterranea, but ended up with a collection of more than 100,000 short pieces. The reason for this is that a great deal of the genome consists of many, nearly identical copies of the same sequence that repeats over and over.
New sequencing methods
To overcome this challenge of an exceptionally repetitive genome, the research groups of Jochen Rink and Eugene Myers at the MPI-CBG utilized Pacific Bioscience’s long-read sequencing technology, operated at the DRESDEN-concept Sequencing Center, a joint operation between the MPI-CBG and the TU Dresden. This relatively new technology can directly “read” contiguous stretches of the genome up to 40,000 base pairs (or “letters”) long. Such long reads are dramatically more effective at bridging repetitive stretches in the genome than the more broadly used 100-500 base pair reads, thus resulting in up to 100-fold improvements in genome assembly statistics over previous assemblies.
Siegfried Schloissnig (HITS) was primarily responsible for developing a novel software system, called “Marvel”, that solves more of the jigsaw puzzle posed by the long-reads than previous such systems, and more efficiently. The assembly of the Schmidtea mediterranea genome involved eight terabytes of data that took the high-performance computing cluster at the HITS three weeks to complete.
Missing genes
But what can scientists actually do with the abundance of genetic information in a genome assembly? One of the surprises in the case of Schmidtea mediterranea was the likely absence of highly conserved genes such as MAD1 and MAD2. Both are present in nearly all other organisms because they fulfil a function in a checkpoint that ensures that both daughter cells get the same number of chromosomes after cell division. Yet despite the MAD1/2 gene loss, planarians retained the checkpoint function. How this is possible is one of the questions that the genome will help to answer. But Jochen Rink and his group are especially excited about using the genome assembly for understanding how planarians manage to regenerate from an arbitrary tissue piece. Rink explains: “We already know some of the genes required for regenerating a head, but now we can also search for the regulatory control sequences that activate the head genes only at the front end of a regenerating piece”. Further, the Rink group has assembled a large collection of planarian species from around the world, many of which have lost the ability to regenerate. “With a powerful toolbox for the assembly of difficult genomes now in place, we hope to soon use genome comparisons to understand why some animals regenerate, while so many do not. At least in the case of flatworms”, summarizes Rink.
Source: MPG