
Diabetes affects more than 400 million individuals worldwide. In what is becoming a paradigm shift, researchers have begun to find that the disease may result in part through pancreatic beta cells losing their functional identity and shutting down their ability to release the blood sugar-lowering hormone, insulin. Researchers from the Max Planck Institute of Immunobiology and Epigenetics in Freiburg find evidence for a new model underpinning this “de-differentiation”. In addition to metabolic stress, Andrew Pospisilik and his team show that breakdown of an epigenetic barrier is required, and indeed sufficient, to drive de-differentiation. Patient data suggest a central role for such impaired epigenetic control in the development of the disease in humans. The new insights, especially relevant for patients sensitive to de-differentiation diagnostically, have strong therapeutic potential.

Pancreatic islets are collections of cells in the pancreas that consist of up to 80 percent of insulin-producing beta cells. An approximately eight-week-old mouse (left) still shows significantly high levels of secreted insulin (magenta) to control blood sugar levels. In comparison, a 25 weeks old mouse (right) with epigenetic dysregulation of the beta cells show markedly limited insulin production. Credit: MPI f. Immunobiology and Epigenetics
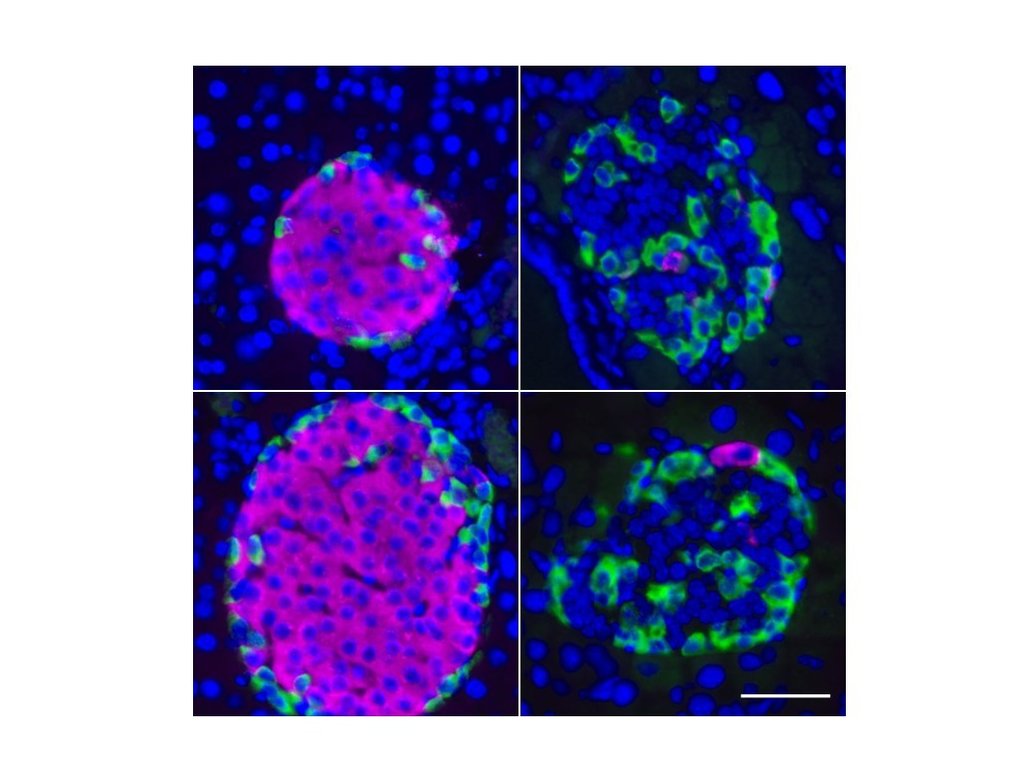{kind=link}
According to the International Diabetes Federation (IDF), diabetes mellitus affects more than 6.5 million people in Germany. With a share of over 95 percent, most patients are suffering from type 2 diabetes, which usually occurs in old age and is associated with obesity and cardiovascular problems. Faulty insulin regulation triggers the widespread disease. When blood sugar levels rise after a meal and insulin is needed quickly in high quantities, the pancreas of patients releases the hormone too slowly leading to dangerously high levels of glucose in the blood.
It has long been thought that reduced insulin production by the pancreas is due to the death of the organ's beta cells that secrete the insulin. However, there has been evidence that beta cells do not die but rather change into a different cell type. Beta cells in patients suffering from type 2 diabetes losing their identities by undergoing a process called de-differentiation. They lose their most specialized functions and revert to a state similar to their immediate developmental precursor, a progenitor-like endocrine cell lacking the ability to secret insulin.
“Metabolic stress has been thought of as the primary trigger of de-differentiation. Here, we show that a second arm is required, namely breakdown of an epigenetic barrier that normally hones beta-cell functional identity. Two independent pathological mechanisms appear to be required. This strong buffer for identity makes sense,” says Andrew Pospisilik, “in humans beta-cells can live upwards of 40 years, so the cells need strong mechanisms to continuously reinforce functional acuity”.
Role of epigenetics in complex diseases
The team around the epigeneticist at the Max Planck Freiburg is driven by the interest in understanding of epigenetic effects in complex diseases such as diabetes, obesity and cancer. They are called complex because they result from a complex genetic predisposition but also significant non-genetic components, often termed ‘environmental influences’. This non-genetic regulation is believed to converge upon chromatin-dependent processes. In our cells DNA is packaged around histone proteins to make this chromatin structure. The packaging of the DNA plays a crucial role in cell type-specific gene regulation, in which genes can be either switched ‘on’ or ‘off’.
“In the end, healthy and de-differentiated beta cells both contain the same DNA. What makes the difference are epigenetic identity barriers that are mediated by modifications of the DNA packaging. In some ways, these processes are like sheet music for an orchestra. They focus and coordinate how and when genes are activated or silenced,” explains Tess Lu, first author of the study.
Chromatin alterations in diabetes
By profiling thousands of beta cells from non-diabetic and type-2 diabetic individuals in mice and humans the team found that two out of about 25 different types of chromatin packaging the DNA, track with beta cell dysfunction: one kind of chromatin was dysregulated specifically in diabetic individuals and another one was surprisingly up-regulated, which is normally supposed to be very silent.
“If you start swapping the sheet music between the instruments of an orchestra, you still get sound, you still get melody, but the music would change dramatically. Similarly in cells, if the genetic programs aren’t correctly coordinated cellular identity changes, and functional specializations fade. Over time this leads to beta cells forgetting who they are and what they are supposed to do,” explains Andrew Pospisilik.
To validate their observations, the researchers triggered these switches to recapitulate the human disease etiology in mice. Animals with this modification were first healthy and developed regular insulin-producing beta cells. But at around middle-age, cells de-differentiated and the animals could not control their blood sugar anymore.
New subtype of type 2 diabetes?
Most interestingly the researchers from Freiburg add a new level of understanding to how we think of de-differentiation in diabetes. Previously thought to be a one-hit process, downstream of metabolic stress or high glucose, the Max Planck team were able to show that a second, epigenetic “failure” is also required, and is indeed sufficient to drive beta-cell de-differentiation and dysfunction.
For the Max Planck researchers, it is a huge step forward in understanding this widespread disease. The findings suggest novel therapeutic strategies at least for type 2 diabetes, but potentially also for type-1. It raises questions whether patient populations may exist that are more sensitive or resistant to the process. “In theory, these epigenetic systems are pharmacologically tractable as any other enzymatic components in a cell. Indeed, such epigenetic therapies are already used in cancer. Targeting epigenetic maintenance of beta cell identity should be actively explored,” says Andrew Pospisilik.
Source: MPG